 Laboratory of Statistical Thermodynamics and Macromolecules
Laboratory of Statistical Thermodynamics and MacromoleculesA number of approaches have been reported in the last years capable of identifying topological constraints and generating ensembles of primitive paths in entangled, multi-chain polymeric systems. In addition to providing predictions for the static (statistical) properties of the underlying entanglement network, these approaches have opened up the way to interfacing atomistic simulation data with reptation, admittedly the most successful phenomenological theory of polymer dynamics for entangled systems. We are developing such a link between atomistic molecular dynamics simulation results and reptation theory by geometrically constructing the effective tube around each primitive chain and then documenting chain motion in terms of a curvilinear diffusion inside the effective tube around the coarse-grained chain contour. The outcome of such a topological and dynamical mapping is the computation of observables quantifying reptation in entangled polymers. A typical example is the function ψ(s,t), namely the probability that a segment s of the primitive chain remains inside the initial tube after time t. We try to utilize this information in order to bring together three different approaches to polymer dynamics (in addition to acquiring reliable experimental data): atomistic simulations, mesoscopic entanglement networks, and tube models. By consistently mapping the results of accurate computer models of polymer structure and dynamics onto theoretical treatments based on phenomenological concepts (that sometimes defy precise definition) on some well-defined model systems, we hope to get a deeper understanding of the predominant relaxation mechanisms in entangled polymers, and thus succeed in our effort to encode this information in the form of suitable (more accurate) constitutive equations.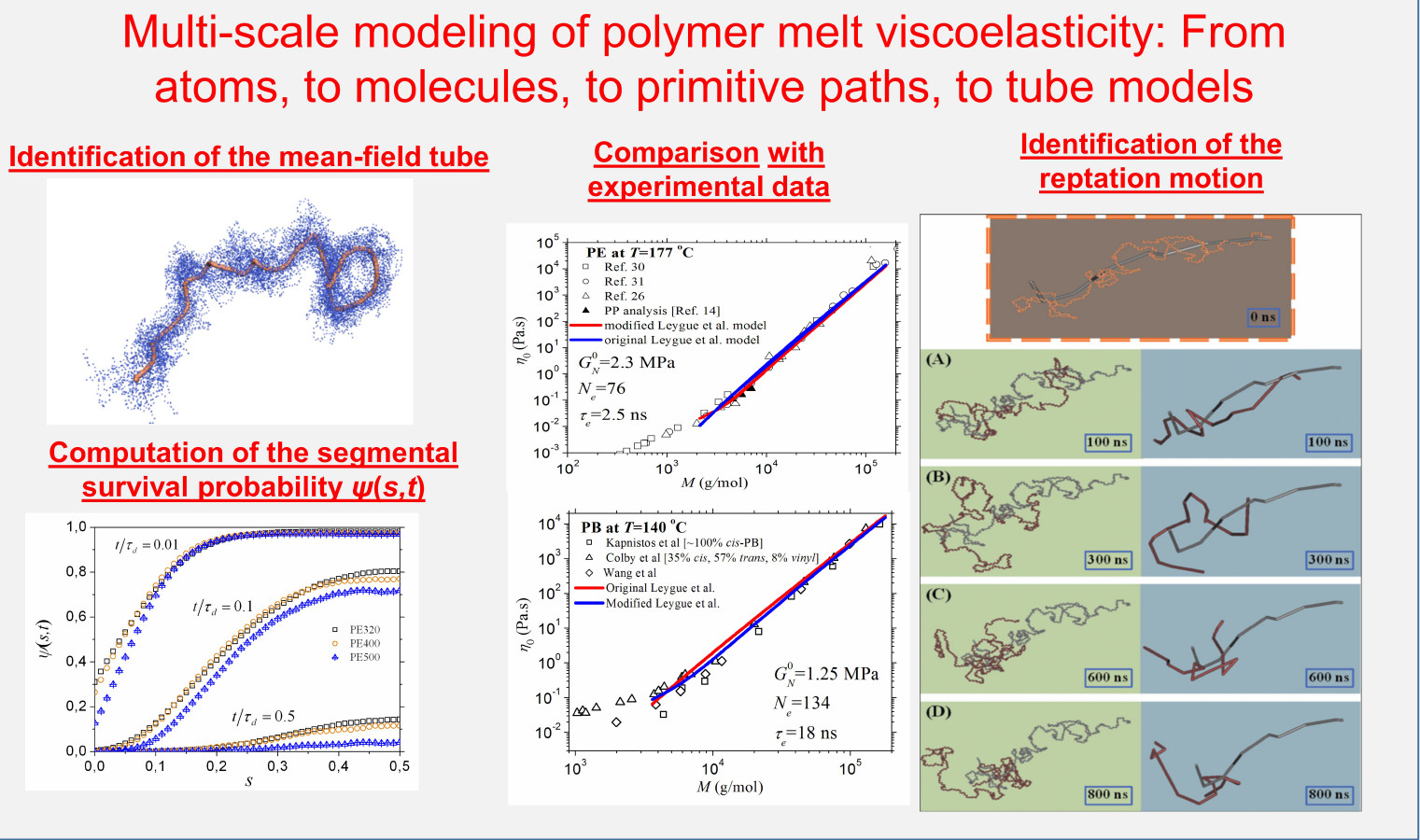
Mapping of atomistic simulation results onto the tube model for the dynamics of entangled polymers